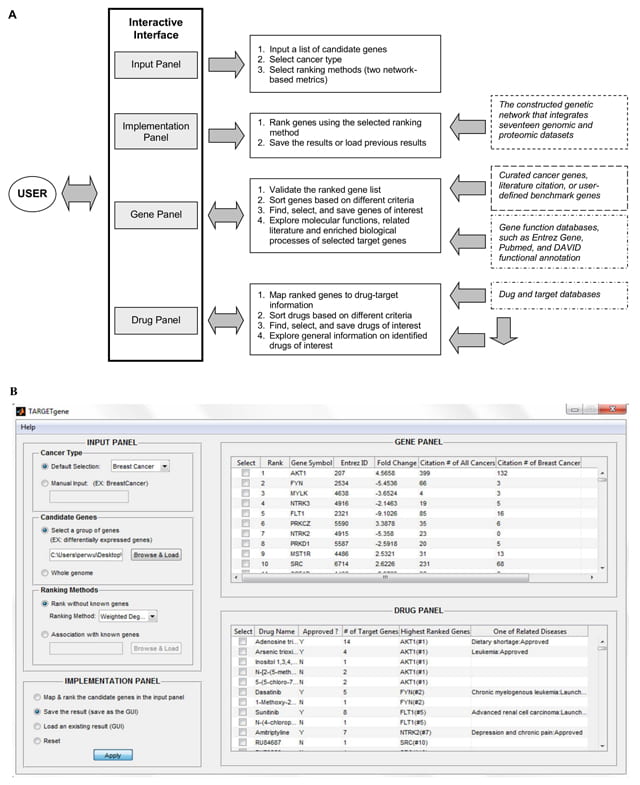
The predictions of TARGETgene are based on a whole-genome genetic network constructed by integrating heterogeneous functional data using RVM-based ensemble model reported by Wu et al., [1]. The nodes in this network represent all genes of the human genome, and the functional association between any two of them is quantified by a gene-pair linkage probability that can reveal the tendency of genes to operate in the same or similar pathways. Based on this constructed gene network, TARGETgene identifies potential therapeutic targets using one of two network-based metrics: 1) hub score, which uses a centrality measure to identify hub genes in a tumor-specific network, or 2) seed gene association score, which quantifies each genes association with known cancer (disease) genes. All the candidate genes are ranked based on their hub score or their seed gene association score. Those highly ranked genes in the prediction are identified as possible important cancer genes and thus potential therapeutic targets. Finally, drug-target information compiled from several drug databases is mapped to candidate genes. Drugs whose target genes are highly ranked in the prediction can be considered as potential therapies.
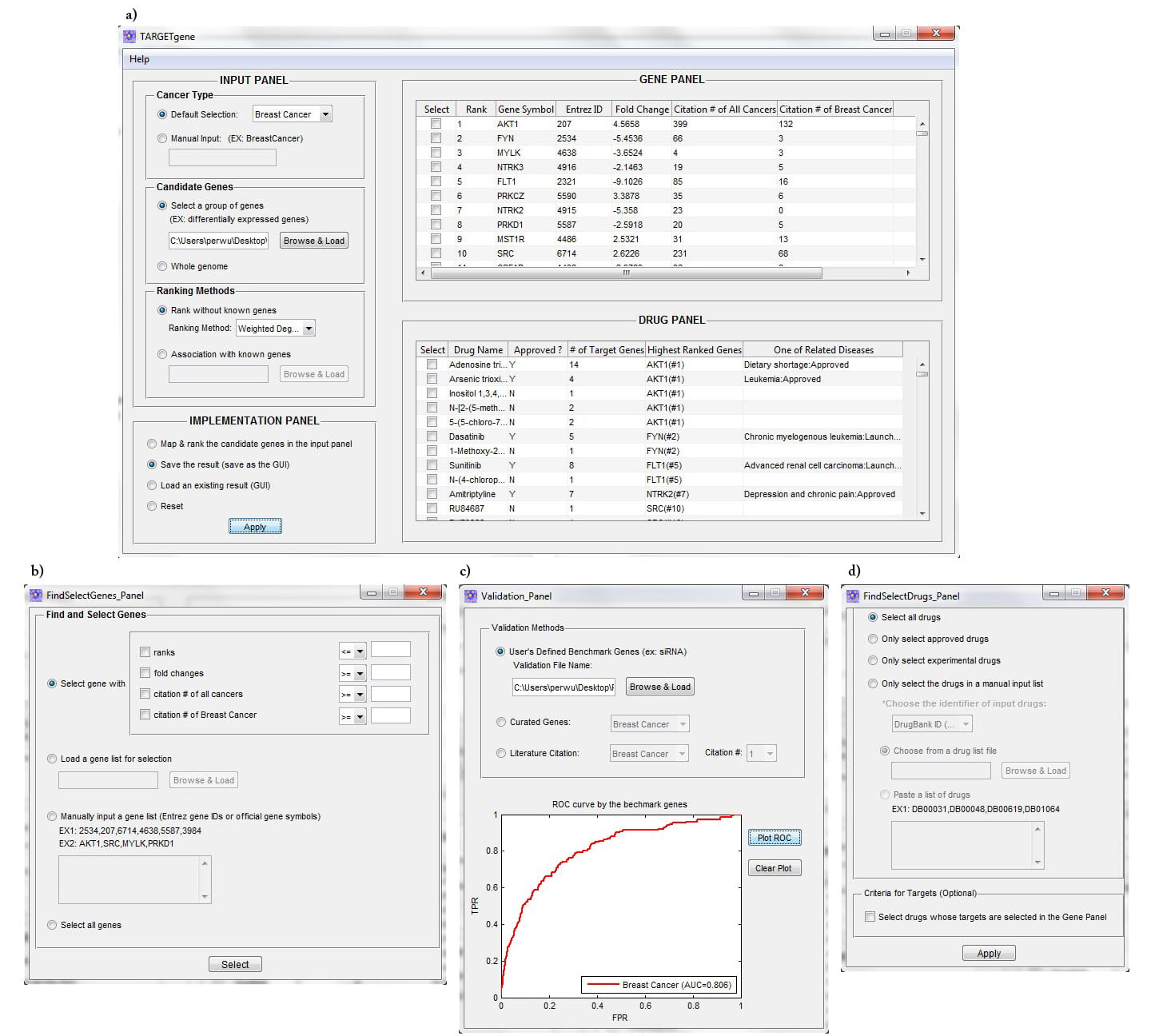
The graphical user interface of TARGETgene consists of four main working panels, including Input, Implementation, Gene, and Drug panels (Figure 1A&B). The Input Panel enables users to define the cancer type (currently: breast cancer, colon cancer, Ewing’s sarcoma, glioblastoma, lung cancer, ovarian cancer, and prostate cancer) and candidate genes, as well as the desired ranking metric (hub score or seed gene association score) as illustrated in Figure 1a. The Implementation Panel allows the user to generate new predictions, save results and load existing results. The Gene Panel lists information on all candidate genes including their rank in predictions, as well as cancer literature citation number. Cancer literature citation information of genes was compiled from Pubmed. Through this panel, the user also can find, select identified target genes (e.g., selecting novel cancer genes), and explore their functions, cited literature as well as enriched biological processes. In addition, TARGETgene enables users to validate their predictions using user-defined benchmark genes (e.g., target genes detected in RNAi screens) and curated cancer genes via this panel. Finally, drug and their target information compiled from several public databases, such as DrugBank [2], PharmGKB [3] and the Therapeutic Target Database [4], is also integrated to TARGETgene for reporting those drugs/compounds that could have action on the targets identified by TARGETgene. The Drug Panel lists generic names, drug types (approved or experimental), number of candidate genes known to be targeted by the identified drugs, highest ranked target gene name, and related diseases of the identified drugs. The list of drugs is ordered by their highest ranked target gene. TARGETgene is also customizable and can generate the list of selected drugs based on the ranks of their targets or drug type. In addition, users can explore more general information on identified drugs of interest through several external links.
File Formats for Input Gene Sets
TARGETgene allows four types of specification formats for input gene lists (a set of candidate genes for prioritize; a set of benchmark genes for validation; a set of seed genes). Users can specify a file that contains a list of differentially expressed genes and their fold changes for ranking. TARGETgene will then rank these genes using genetic network-based approaches. The file should be a tab-delimited text file with no header and two columns: Entrez gene ID (or official gene symbol) and fold changes of the gene expression.

Users can also specify a file that only contains a set of genes. It should be a text file with no header and one column: Entrez gene ID (or official gene symbol). For example, users may rank a set of genes with mutations or copy number changes in cancer to identify driver genes in oncogenesis. In addition, users may also use these two formats to input a set of seed genes for functional association or a set of genes for validation of predictions.

References
- Wu, C.C. et al. (2010) Prediction of Human Functional Genetic Networks from Heterogeneous Data Using RVM-Based Ensemble Learning. Bioinformatics 26(6): 807-813.
- Wishart, D.S. et al. (2008) Drugbank: a knowledge base for drugs, drug actions and drug targets. Nucleic Acids Res. 36(Database issue): D901-906.
- Hodge,A.E. et al. (2007) The PharmGKB: integration, aggregation, and annotation of pharmacogenomic data and knowledge. Clin Pharmacol Ther. 81(1): 21-24.
- Zhu,F. et al. (2009) Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 38(Database issue): D787-791.
File Formats for Input Gene Sets
TARGETgene allows four types of specification formats for input gene lists (a set of candidate genes for prioritize; a set of benchmark genes for validation; a set of seed genes). Users can specify a file that contains a list of differentially expressed genes and their fold changes for ranking. TARGETgene will then rank these genes using genetic network-based approaches. The file should be a tab-delimited text file with no header and two columns: Entrez gene ID (or official gene symbol) and fold changes of the gene expression.
Users can also specify a file that only contains a set of genes. It should be a text file with no header and one column: Entrez gene ID (or official gene symbol). For example, users may rank a set of genes with mutations or copy number changes in cancer to identify driver genes in oncogenesis. In addition, users may also use these two formats to input a set of seed genes for functional association or a set of genes for validation of predictions.
References
- Wu, C.C. et al. (2010) Prediction of Human Functional Genetic Networks from Heterogeneous Data Using RVM-Based Ensemble Learning. Bioinformatics 26(6): 807-813.
- Wishart, D.S. et al. (2008) Drugbank: a knowledge base for drugs, drug actions and drug targets. Nucleic Acids Res. 36(Database issue): D901-906.
- Hodge,A.E. et al. (2007) The PharmGKB: integration, aggregation, and annotation of pharmacogenomic data and knowledge. Clin Pharmacol Ther. 81(1): 21-24.
- Zhu,F. et al. (2009) Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 38(Database issue): D787-791.
–>
Latest Posts About TARGETgene
- TARGETgene 2.0betaA fusion gene that results from chromosome rearrangements is a hybrid gene formed from distinct genes, either in the same chromosome or in different chromosomes. Many recurring gene fusions, such as BCR-ABL in chronic myelogenous leukemia, EWS-FIL1 in Ewing’s Sarcoma, and TMPRSS2-ERG in prostate cancer, play important roles in cancer progression. As such, they have…Continue Reading TARGETgene 2.0beta
- Release of TARGETgeneThe vast array of in silico resources currently available in the life sciences research offers the possibility of aiding the drug discovery process. Core Project 1 of the BMSR has developed TARGETgene to exploit these resources and to allow the identification of potential therapeutic targets and drugs in cancer using genetic network-based approaches. TARGETgene is…Continue Reading Release of TARGETgene